-



高精度計算化學平臺

ID4Gibbs?基于量子力學、分子力學、統計力學等物理原理,構建了自研分子力場XFF、結合模式預測XPose和親和力預測XFEP流程,從精度、效率和通量上有力地支持FIC、BIC、FF等業務項目的分子設計、評估和優化。
-
XFF-自研高精度分子力場
- 精度高
根據>50個真實管線應用反饋,對力場和算法進行了多輪迭代;
- 海量訓練數據
已完成數億核時,百萬量級的量子化學計算以及數千次分子動力學模擬;
- 全面覆蓋類藥分子化學空間
多樣化、針對性設計的內部訓練集;
- 云平臺部署
自動進行參數修正,可為客戶化學空間定制專有力場開發。
-
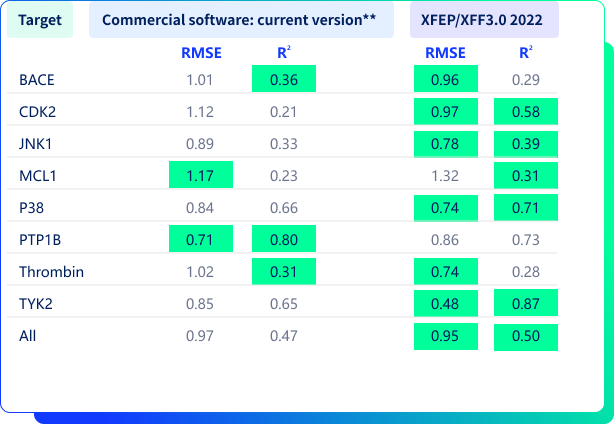
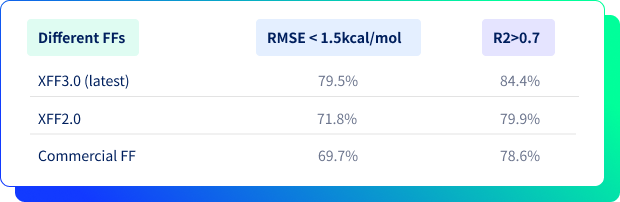
親和力預測對比結果
QM vs MM構象能對比(>10萬分子集)
-
XPose攻克“不可成藥靶點”
- 精度高
對接結果達到原子級精度(RMSD<1.0 Angstrom);
- 性能強
以實際項目體系進行訓練和測試,更貼近實際項目需求;
- 柔性對接
對于小分子結合誘導氨基酸側鏈構象變化的體系,能夠準確預測小分子的結合模式,以及小分子結合后的位置和構象。
-
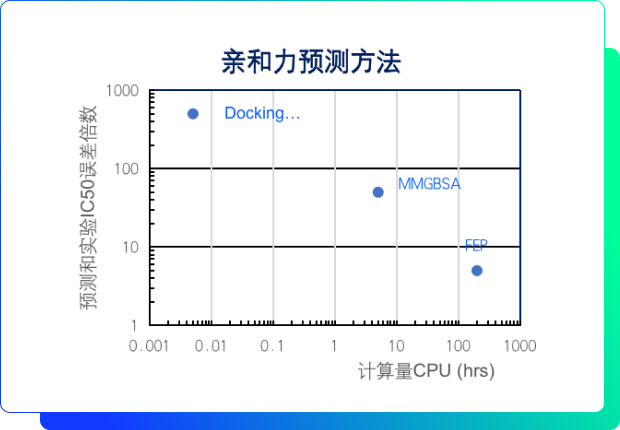
XFEP高通量親和力評估
- 精度高
預測活性誤差在1個數量級以內;
- 效率高
可以在一周之內精確計算出5000-10000個小分子藥物候選化合物和靶點的結合自由能;
- 通量高
其他同類商業方法的10-100倍;
- 功能全
涵蓋藥物設計大量實際場景需求,如耐藥性和選擇性預測,PROTAC分子、共價結合化合物、全新蛋白及短肽藥物設計,基于片段/結構的骨架篩選、理性骨架設計和優化、官能團篩選、化合物結合構象驗證等。